ANGIOEDEMA SEM URTICAS – TIPOS
ANGIOEDEMA HEREDITÁRIO
É uma doença genética rara causada pela alteração de uma proteína denominada inibidor de C1 (C1-INH), que pode ser hereditária ou causada por uma mutação.
A característica principal dessa doença é o aparecimento de grandes inchaços de maneira recorrente durante a vida. Esse angioedema afeta principalmente a face, mãos, pés, genitais, laringe e intestinos.
Os inchaços causam grande incômodo estético por serem deformantes e muitas vezes dolorosos. Quando acometem o intestino, podem causar dor abdominal, diarreia e até vômitos. O maior problema ocorre quando acomete a laringe, que é uma parte das vias respiratórias, pois pode levar a um quadro de asfixia e até morte se não for tratado rapidamente.
As crises de angioedema hereditário ocorrem espontaneamente ou em decorrência de infecções, cirurgias, traumas, estresse, menstruação.
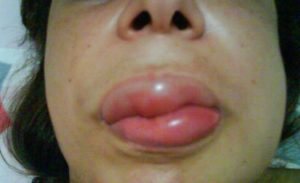
ANGIOEDEMA ADQUIRIDO
É uma forma muito rara de angioedema sem urticas que ocorre pela deficiência de uma proteína denominada inibidor de C1 (C1-INH), e pode ocorrer em portadores de doenças linfoproliferativas (como, linfomas) ou doenças autoimunes.
Os sintomas são semelhantes ao do angioedema hereditário.
ANGIOEDEMA PELO INIBIDOR DA ENZIMA DE CONVERSÃO (IECA)
A sigla IECA (inibidor da enzima da conversão da angiotensina) é a abreviação de um grupo de medicamentos muito utilizados para o controle da hipertensão e da insuficiência cardíaca. São aqueles medicamentos cujo nome termina em “pril”, como por exemplo captopril, enalapril, fosinopril, lisinopril e quinapril.
Até 3% dos pacientes que tomam estes remédios podem ter crises repetidas de angioedema por aumento da bradicinina. Estas crises podem começar logo após o início do tratamento, após meses ou até anos. Isso torna o diagnóstico difícil. Nesse tipo de angioedema, não ocorre alteração no inibidor de C1.
Acomete principalmente a região facial, lábios e língua, podendo levar a asfixia por fechamento das vias aéreas superiores. Os inchaços são muito intensos e duradouros.
Bibliografia:
Allergy 2014 May;69(5):602-16
Int J Emerg Med. 2012 Nov 6;5(1):39
J Am Acad Dermatol. 2011 Oct;65(4):843-50
Clin Rev Allergy Immunol. 2016 Oct;51(2):183-92



